MIT Lincoln Lab and Tuple Achieve Immense Scalability in SARS-CoV-2 Molecular Modeling with High-Performance Computing

In a paper released recently in Frontiers in Bioinformatics, researchers from MIT Lincoln Lab and Tuple share their results of a large-scale, in silico study of coronavirus nucleocapsid interactivity with human cytokines. This study aimed to better understand the variability in the human immune response among infections with SARS-CoV-2 variants. These findings offer valuable insights into the molecular mechanisms underlying viral pathogenesis and may guide the future development of targeted interventions. Also, the computational framework created for this study will enable future molecular modeling at-scale.
In this study, researchers predicted the binding affinities between 64 human cytokines against 17 coronavirus nucleocapsid proteins. This resulted in 1,088 complexes that were generated and analyzed. The team also compared the docking results between the deep learning-based AlphaFold2-Multimer and the semi-physicochemical-based HADDOCK 2.4 for protein-protein interactions.
Achieving Grand Scale
Since there were 1,088 experiments to perform and given that both AlphaFold2-Multimer and HADDOCK jobs take hours to complete, distributing the processing on the MIT SuperCloud high-performance computing (HPC) environment allowed for incredible throughput of the experiments.

AlphaFold2-Multimer Benchmarks
On nodes with NVIDIA Volta V100 GPUs (5,120 CUDA cores, 640 tensor cores), jobs took 493 ± 21 minutes of walltime per multimer run.
A subset of jobs were tested on nodes with NVIDIA Ampere A100 GPUs (6,912 CUDA cores, 432 tensor cores) and the mean walltime decreased to 378 ± 80 minutes. This is presumably due to the increased number of CUDA cores being utilized.
The 1,088 experiments were run on 36 nodes with NVIDIA Volta V100 GPUs, completing the entire set of experiments in ~12 days.
HADDOCK Benchmarks
For HADDOCK, two different types of HPC nodes were used to test the effect of core quantity on the speed of jobs.
- On Intel Xeon Phi 7210 (1.5GHz) 64 core cluster nodes, the walltime was 707 ± 40 minutes.
- On Intel Xeon Platinum 8260 (3.9GHz) 48 core cluster nodes, the walltime decreased to 133 ± 92 minutes.
Despite the Phi 7210 nodes having more cores, the Platinum 8260 nodes, with the faster clockspeed, provided superior throughput.
The 1,088 experiments were run on 64 Xeon Platinum 8260 nodes, completing the entire set of experiments in ~36 hours.
Run times varied, depending on the cytokine and number of surface residues selected against which to dock. Also, cytokines with multiple chains (IL-12p70, IL-23, IL-27, and IL-35) took approximately three times as long to dock.
Containerization is Key
Both AlphaFold2-Multimer and HADDOCK have lots of dependencies that may make them challenging to run. Using Docker and Singularity, those dependencies and configuration steps can be packaged up in a container, making these tools much more portable and usable in a cluster environment.
Containerization is important no matter if you’re using clusters in the cloud, such as Azure Kubernetes Service, or on-prem, such as on a research HPC environment like the MIT SuperCloud.
The containers used in this study are open-source and provided here:
- AlphaFold2 Singularity Container: https://github.com/mit-ll/AlphaFold
- HADDOCK Docker Container: https://github.com/colbyford/HADDOCKer
Study Findings, In Summary…
Across the 1,088 complexes (and the 2 docking systems tested), there were multiple cytokines with consistently strong binding across all 17 coronavirus N proteins. Of interest, CCL22, IL-18BP, CXCL12β, and others.
CCL28, CXCL10, CXCL11, and CXCL14 from the HADDOCK predictions show that the binding affinity worsens as the genetic distance of the N protein increases from wild type SARS-CoV-2. Also, some cytokines, such as CCL27, CXCL12α, TNFα, IL-6, and IFNλ1, show improved binding in newer SARS-CoV-2 variants. These findings pose interesting questions around the role these cytokines play in the human immune response to COVID-19 and why the virus may be mutating to become more/less fit at binding to certain immunoproteins.
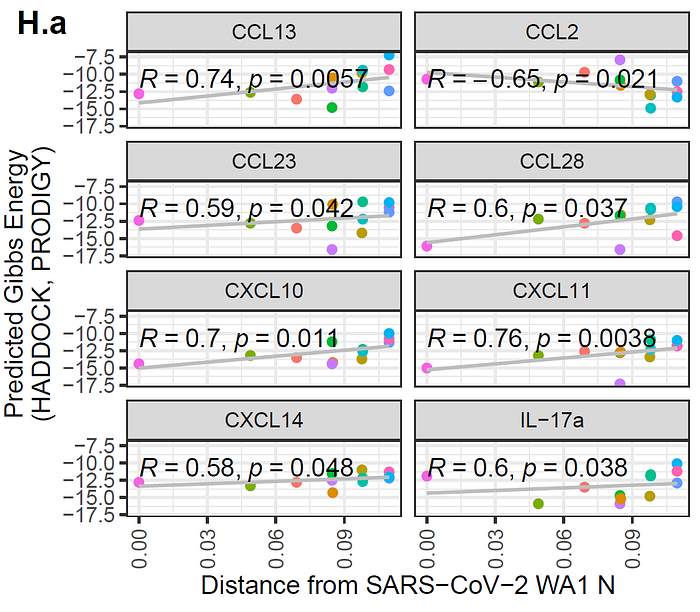
Comparing AlphaFold2-Multimer and HADDOCK
For some cytokines, there are marked differences in the binding predictions between AlphaFold2-Multimer and HADDOCK. For example, in CXCL12β below, the overall binding site is similar, but the interfacing residues are different. Thus, some empirical studies are needed to adjudicate the predictions presented in this work.
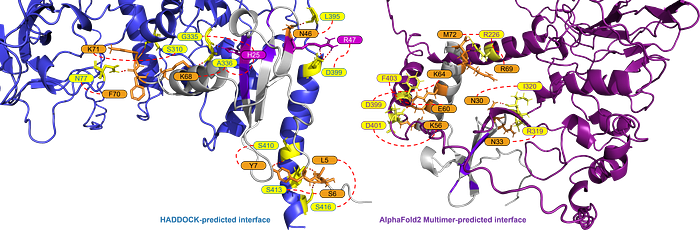
(polar contacts within 3.0Å)
Overall, this study showcases the utility of computational modeling in understanding the interaction between proteins and how pathogens interact with the human immune system. Plus, the scalability demonstrated here allows for future large, in silico studies in drug design, infectious disease research, immunology, and beyond!
- Read the full paper here: https://www.frontiersin.org/articles/10.3389/fbinf.2024.1397968/full
If you’re interested in working with Tuple on a similar project, please reach out to contact@tuple.xyz and let us know you read this case study!
Stay Curious…
Disclaimer
DISTRIBUTION STATEMENT A. Approved for public release. Distribution is unlimited.
This material is based upon work supported by the Department of the Air Force under Air Force Contract No. FA8702–15-D-0001. Any opinions, findings, conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the Department of the Air Force.
